Choose timezone
Your profile timezone:

Quality by Design (QbD) plays an important role in the modern industry. Often, it goes hand in hand with technological solutions that secure the supervision and stability of the processes in the design space (DS), such as spectroscopy, namely near-infrared, Raman, fluorescence, or UV, collectively called Process Analytical Technology (PAT). QbD and PAT initiatives have benefited from concepts, methodologies and tools arising from different corners of the data-driven sciences, such as statistics, chemometrics, and machine learning. The integration of first principles and existing knowledge with empirical modelling, called grey modelling, is another area of current active research that may bring important contributions to QbD/PAT. Inevitably, Artificial Intelligence will also play a role in the future of these fields, but which exactly is an open question. Therefore, it is both important and opportune to assess how these different approaches can further contribute to improving or reinventing QbD and PAT initiatives, either isolated or cooperatively. The main goal of the ENBIS Spring Meeting 2025 is to foster high-level discussions on these and other related topics.
All stakeholders, from graduate students to professionals, from researchers to industrial managers, are invited to participate actively. We welcome contributions in the following areas (the list is not exhaustive), with a focus on their application in industry in the scope of QbD/PAT:

Contact information
For any question about the meeting venue and scientific programme, registration and paper submission, feel free to contact the ENBIS Permanent Office : office@enbis.org.
Local Organizing Committee:
Programme Committee:

Antonio Del Rio Chanona (Imperial College, UK)
LLM and human-in-the-loop Bayesian optimization for chemical experiments
Bayesian optimization has proven effective for optimizing expensive-to-evaluate functions in Chemical Engineering. However, valuable physical insights from domain experts are often overlooked. This article introduces a collaborative Bayesian optimization approach that integrates both human expertise and large language models (LLMs) into the data-driven decision-making process. By combining high-throughput Bayesian optimization with discrete decision theory, experts and LLMs collaboratively influence the selection of experiments via a human-LLM-in-the-loop discrete choice mechanism. We propose a multi-objective approach to generate a diverse set of high-utility and distinct solutions, from which the expert, supported by an LLM, selects the preferred solution for evaluation at each iteration. The LLM assists in interpreting complex model outputs, suggesting experimental strategies, and mitigating cognitive biases, thereby augmenting human decision-making while maintaining interpretability and accountability. Our methodology retains the advantages of Bayesian optimization while incorporating expert knowledge and AI-driven guidance. The approach is demonstrated across various case studies, including bioprocess optimization and reactor geometry design, showing that even with an uninformed practitioner, the algorithm recovers the regret of standard Bayesian optimization. By including continuous expert-LLM interaction, the proposed method enables faster convergence, improved decision-making, and enhanced accountability for Bayesian optimization in engineering systems.

Establishing Multivariate Specification Regions for Incoming Raw Materials – a QbD approach
Establishing multivariate specification regions for selecting raw material lots entering a customer’s plant is crucial for ensuring smooth operations and consistently achieving final product quality targets. Moreover, these regions guide the selection of suppliers. By meeting these specifications, suppliers contribute to customer satisfaction, which can, in turn, enhance market share. Latent Variable Methods, such as Partial Least Squares regression (PLS) and the more recent Sequential Multi-block PLS (SMB-PLS), have proven to be effective data-driven approaches for defining multivariate specification regions. These methods model the relationships between raw material properties (Critical Material Attributes – CMA), process variables (Critical Process Parameters – CPP), and final product quality (Critical Quality Attributes – CQA), enabling the identification of a lower-dimensional latent variable subspace that captures quality-relevant variations introduced by raw materials and process conditions. This subspace is central to the methodology.
Within this latent variable space, several statistical limits are defined to ensure final product quality and guarantee data compliance with the latent variable model, collectively forming the multivariate specification region. After providing historical context on the early development of these methods, this presentation will cover several key topics. These include the data requirements for constructing latent variable models and how to organize them into distinct blocks. The techniques used to define the limits (in terms of shape and size) within the latent variable subspace—via direct mapping and latent variable model inversion—will be discussed, along with methods for addressing uncertainties. Particular attention will be given to how process variations generate different scenarios and approaches for establishing meaningful specification regions. Case studies using both simulated and industrial data will illustrate these methods. It will be demonstrated that the proposed framework aligns with the principles of Quality by Design (QbD), with notable similarities to the Design Space (DS) concept. The presentation will conclude by exploring potential future developments, including the establishment of multivariate process capability indices based on specification regions.
Bayesian optimization has proven effective for optimizing expensive-to-evaluate functions in Chemical Engineering. However, valuable physical insights from domain experts are often overlooked. This article introduces a collaborative Bayesian optimization approach that integrates both human expertise and large language models (LLMs) into the data-driven decision-making process. By combining high-throughput Bayesian optimization with discrete decision theory, experts and LLMs collaboratively influence the selection of experiments via a human-LLM-in-the-loop discrete choice mechanism. We propose a multi-objective approach to generate a diverse set of high-utility and distinct solutions, from which the expert, supported by an LLM, selects the preferred solution for evaluation at each iteration. The LLM assists in interpreting complex model outputs, suggesting experimental strategies, and mitigating cognitive biases, thereby augmenting human decision-making while maintaining interpretability and accountability. Our methodology retains the advantages of Bayesian optimization while incorporating expert knowledge and AI-driven guidance. The approach is demonstrated across various case studies, including bioprocess optimization and reactor geometry design, showing that even with an uninformed practitioner, the algorithm recovers the regret of standard Bayesian optimization. By including continuous expert-LLM interaction, the proposed method enables faster convergence, improved decision-making, and enhanced accountability for Bayesian optimization in engineering systems.
Design of experiments for process scale-up can be described as a double-edged sword for the pharmaceutical industry: intensification of experiments expands the knowledge of the process (uncertainty reduction) but increases resource expenditure. On the other hand, moving forward without enough process understanding is the first stone in a path of deviations, lack of quality, and even safety concerns.
In the past years, Bayesian sampling methodologies have surfaced to incorporate uncertainty and lead to better guided risk/optimal decision making in terms of process conditions, and reduction of required experiments. Utilizing Bayesian sampling for design space offers several significant advantages: First, it allows for the incorporation of prior knowledge, leading to more informed and efficient experimental designs [1]. Secondly, by continuously updating beliefs with new data, Bayesian sampling enables a dynamic and adaptive approach, enhancing the accuracy and reliability of results. This method also provides a rigorous framework for quantifying uncertainty, ensuring robust decision-making even in complex scenarios [2]. Additionally, Bayesian sampling can effectively identify the probability space with reduced experimental work, leading to an earlier definition of a Normalized Operating Range (NOR) within a scale-up approach to a pharmaceutical process.
In this work, a batch gas generating process with 10 different reactions occurring (reagents, products and by-products) is evaluated with the proposed Bayesian Design Space [1], with different process parameters defined (time, temperature, reagent “A” initial concentration and reagent/solvent “B” initial concentration) and consumption CQA’s required for the same process. The results showed that a reduced amount of experiment (less than 6) were required to achieve an acceptable NOR for the process, and the outcome allowed for a safe transfer to a higher volume unit (manufacturing) with all safety and quality requirements achieved.
[1] – Kusumo, K. et al., “Bayesian Approach to Probabilistic Design Space Characterization: A Nested Sampling Strategy”, I&EC research, 2019
[2] – Kennedy, P. et al., “Nested Sampling Strategy for Bayesian Design Space Characterization”, Comp. Aided Chem. Eng., 2020
Capability indexes can be used to estimate how likely a given supplier of raw materials is to meet customer's requirements for these raw materials. It is therefore usually used by a customer operating a process as a criterion for selecting raw material suppliers. However, both univariate and multivariate capability indexes provided so far in the literature assume that the specifications are regions defined in the original space of raw material properties without considering their relationships with the Critical Quality Attributes (CQAs) of the manufactured product. For that reason, these specifications may become meaningless, increasing the costs in the acquisition of raw material lots. Alternatively, calculating capability indexes directly within the CQA space would require manufacturing products from raw material lots, without knowing in advance if the product manufactured is going to be good or bad, with the subsequent potential associated costs. Therefore, the supplier assessment could be considered as a Capability by Testing approach.
We present a novel Latent Space-based Multivariate Capability Index (LSb-MCpk) that resolves these two challenges [1]. The most remarkable advantage of the proposed LSb-MCpk is that it is not exclusively defined either in the multivariate raw material space or in the CQA space of the product manufactured, but in the latent space connecting both. This advantage is key to establishing the so-called Capability by Design aligned with the Quality by Design initiative. Indeed, the LSb-MCpk quantifies the ability of each supplier of a particular raw material to produce a certain percentage of the final product with assurance of quality which could be used for real-time release. This information can be obtained at the reception of the supplier´s raw materials, before producing a single unit of the product. In addition, diagnosing assignable causes can be carried out when the lots of the supplier raw materials do not respect the correlation structure from the past (by using the Squared Prediction Error contribution plots), or when the supplier cannot consistently operate within the design space (by using the score contribution plots). The proposed LSb-MCpk is based on Partial Least Squares (PLS) regression, and it is illustrated using data from an industrial study.
[1] J. Borràs-Ferrís, C. Duchesne, and A. Ferrer, Chemom. Intell. Lab. Syst., 258, 2025.
In process robustness studies, experimenters are interested in comparing the responses at different locations within the normal operating ranges of the process parameters to the response at the target operating condition. Small differences in the responses imply that the manufacturing process is not affected by the expected fluctuations in the process parameters, indicating its robustness. In this presentation, we propose an optimal design criterion, named the generalized integrated variance for differences (GID) criterion, to set up experiments for robustness studies. GID-optimal designs have broad applications, particularly in pharmaceutical product development and manufacturing. We show that GID-optimal designs have better predictive performances than other commonly used designs for robustness studies, especially when the target operating condition is not located at the center of the experimental region. In some situations that we encountered, the alternative designs typically used are roughly only 50% as efficient as GID-optimal designs. We will demonstrate the advantages of tailor-made GID-optimal designs through an application to a manufacturing process robustness study of the Rotarix liquid vaccine.
The Quality by Design (QbD) approach has been widely adopted in the development of both novel and generic pharmaceutical formulations1. Extending these principles to the analytical domain, Analytical Quality by Design (AQbD) has emerged as a structured framework for optimizing analytical methodologies2. The aim of the present work was to outline a comprehensive framework for development of a texture analyzer-based mucoadhesion method integrated with rheological analysis for the evaluation of ointment formulations3. The ATP was carefully defined for the mucoadhesion method, taking into account both preliminary studies and method requirements. The AQbD approach facilitated the identification of key sources of variability affecting Critical Method Variables (CMVs). Using this methodological framework, critical analytical attributes (CAAs) such as peak force (adhesiveness), work of adhesion (the area under the force/distance curve), and debonding distance were systematically investigated. Optimal conditions were determined using response surface methodology within the Method Operable Design Region (MODR). The study implemented a three-factor, three-level Box-Behnken design (BBD), a widely accepted design of experiments (DoE) approach for exploring quadratic response surfaces and developing second-order polynomial models. The determination coefficients showed that the quadratic model effectively represented these response variables, confirming its predictive power. The root mean square error values for peak force (0.25) and work of adhesion (1.54) indicate minimal deviation between observed and predicted values. However, the model showed less robustness for the debonding distance response, with an R² of 0.24, explaining only 24% of variability. The RMSE of 44.63 indicates high variability, but remains within an acceptable range for this response parameter. The final optimized conditions—2 N applied force, 60 seconds contact time, and 0.05 N trigger force—were established to ensure compliance with the predefined ATP. The incorporation of mucin dispersion into the method showed significant rheological synergism, validating the robustness of the optimized method. Furthermore, compliance with ICH guidelines confirmed the method's reproducibility and reliability4. Overall, this study demonstrates the successful application of AQbD principles in the development of a mucoadhesion assessment method that integrates texture analysis and rheology to optimize the evaluation of ointment formulations. The use of a Box-Behnken design allowed for systematic optimization, ensuring robust, reproducible, and analytically sound results, thereby strengthening the credibility of AQbD-driven analytical methods.
Acknowledgements
Lucas Chiarentin acknowledges the research grants PD/BDE/150717/2020 and COVID/BDE/153661/2024 assigned by FCT (Fundação para a Ciência e a Tecnologia) and Laboratórios Basi, from Drugs R&D Doctoral Program. The authors also thank UCQFarma for making available the Haake MARS 60 Rheometers (Thermo Scientific, Karlsruhe, Germany). Moreover, we also acknowledged Coimbra Chemistry Center (CQC), supported by FCT, through the project UID/QUI/00313/2020.
References
1.Thorsten, V. et al. Analytical Quality by Design, Life Cycle Management, and Method Control. AAPS PharmSciTech 24, 1–21 (2022).
2.Chiarentin, L. et al. Drilling into “Quality by Design” Approach for Analytical Methods. Crit. Rev. Anal. Chem. 0, 1–42 (2023).
3.Chiarentin, L., Moura, V. & Vitorino, C. Mucoadhesion and rheology characterization in topical semisolid formulations: An AQbD-driven case study. Int. J. Pharm. 673, 125389 (2025).
4.Analytical procedure development - ICH (Q14). at https://database.ich.org/sites/default/files/ICH_Q14_Document_Step2_Guideline_2022_0324.pdf (2024).
Introduction: As Steve Jobs famously stated, "You can't connect the dots looking forward; you can only connect them looking backward." This reflective insight resonates perfectly in this work. Adopt a Retrospective Quality by Design (rQbD) perspective and connect the dots of past manufacturing experiences to drive continuous improvement and innovation in legacy drug products and their processes. This industrial case study demonstrates the application of Retrospective QbD to improve process understanding, robustness, product quality, and regulatory compliance, as well as highlights its potential to foster industrial innovation, ultimately creating a more adaptive and resilient landscape for pharmaceutical manufacturing.
Method: The rQbD was conducted at a pharmaceutical manufacturing facility. Key steps involved: (1) Access the available information on process development, the history of legacy product manufacturing, and feedback from technicians (2) Define the Quality Target Product Profile (QTPP) (3) Identify Critical Quality Attributes (CQAs) (4) Collection and pre-treatment of raw material and manufacturing data (5) Collect additional data through complementary follow-up experiences (6) Identify Critical Material Attributes (CMAs) and Critical Process Parameters (CPPs) using a risk-based approach based on multivariate data analytics. During step (6), it was possible to identify potentially interesting associations and trends in the collected historical manufacturing data, along with additional generated data.
Results: By obtaining CMAs and CPPs through a risk approach based on historical data, it became possible to gain a clearer understanding of process and product variability. This will enable swifter adjustments with fewer instances of "trial and error" to process parameters through the obtained models while also minimizing downtime and reprocessing, thereby enhancing manufacturing efficiency. The regulatory flexibility within an industrial context that could also be facilitated by incorporating the proposed rQbD framework into the Common Technical Dossier (CTD) of drug products may represent another significant potential outcome of this work.
Conclusions: The findings underscore the benefits of the Retrospective QbD approach in optimising the manufacturing processes of legacy products, driving continuous improvement, and ensuring high-quality pharmaceutical production. This approach not only offers a valuable pathway for the pharmaceutical industry to adapt to evolving technological advancements within Pharma 4.0 and regulatory landscapes, but also emphasizes the industry's crucial role in this adaptation.
Process analytic technologies (PAT) are routinely used to rapidly assess quality properties in many industrial sectors. The performance of PAT-based models is, however, highly related to their ability to pre-process the spectra and select key wavebands. Amongst the modeling methodologies for PAT, partial least squares (PLS) (Wold, Sjöström and Eriksson, 2001) and interval partial least squares (iPLS) (Nørgaard et al., 2000) models coupled with well-known chemometric pre-processing approaches are the most widespread due to their ease of use and interpretability. As an alternative to classical pre-processing approaches, wavelet transforms (Mallat, 1989) provide a fast framework for feature extraction by convulsion of fixed filters with the original signal.
The proposed Multiscale interval Partial Least Squares (MS-iPLS) methodology aims to combine the ability of wavelet transforms for feature extraction with those of iPLS for feature selection. To achieve this, MS-iPLS makes use of wavelet transforms to decompose the spectrum into wavelet coefficients at different time-frequency scales, and, afterward, the relevant wavelet coefficients are selected using either Forward addition or Backward elimination algorithms for iPLS. As the wavelet filters are linear, the MS-iPLS model can also be equivalently expressed in the original spectral domain, and thus, the standard PLS approaches can be applied for the sake of interpretability and feature analysis.
In this study, 10 MS-iPLS models variants were constructed using five types of wavelet transforms and two iPLS selection algorithms and compared against 27 PLS benchmarks variants using different chemometric pre-processing and interval selection algorithms. The models were compared in two case studies, addressing a regression problem and a classification problem with real data.
The results show that MS-iPLS models can either match or overcome the performance of the PLS benchmark models. For the regression problem, the PLS benchmark models were able to attain the lowest root mean squared error (RMSE), but their performance range was also wider, from an average RMSE of 0.11 (best model) to 2.46 (worst model), with most models being on the lower end. In contrast, the MS-iPLS models were consistently on the upper end, with an average RMSE ranging from 0.13 (best model) to 0.50 (worst model).
In the classification problem, MS-iPLS attained the best performance with an average accuracy of 92.7%, while the best PLS benchmark model had an average accuracy of 89.0%.
Similarly to the PLS benchmarks models, MS-iPLS still requires an exhaustive search for the optimal wavelet transform for each case study. However, with MS-iPLS the number of models to explore was significantly reduced (by a factor of 3, i.e., 1/3) without compromising on performance or interpretability.
References
Mallat, S.G. (1989) IEEE Transactions on Pattern Analysis and Machine Intelligence, 11(7), pp. 674–693.
Nørgaard, L., Saudland, A., Wagner, J., Nielsen, J.P., Munck, L. and Engelsen, S.B. (2000) Applied Spectroscopy, 54(3), pp. 413–419.
Wold, S., Sjöström, M. and Eriksson, L. (2001) Chemometrics and Intelligent Laboratory Systems, 58, pp. 109–130.
In the pharmaceutical industry, drug solubility is a critical quality attribute. For example, drug solubility in organic solvents mixtures is usually screened in drug development to select the best solvent system for crystallization in such a way as to design the manufacturing process. Solubility is also important in the final product because it has a direct impact on the way the drug is absorbed by the patient. Despite the different challenges posed by different application domains, we propose digital models to predict solubility both in organic solvent mixtures for crystallization process development and in gastrointestinal fluids for oral drug absorption.
The first machine-learning model predicts the solubility of drugs in mixtures of organic solvents commonly used in crystallization processes. The new proposed method identifies solvents using molecular descriptors based on UNIFAC subgroups. Solubility prediction is achieved as a function of temperature, solvent functional subgroups, and mixture composition employing Partial Least-Squares (PLS) regression.
The second approach addresses the challenge of predicting drug solubility in the human gastrointestinal tract, a key factor influencing bioavailability for evaluating its therapeutic efficacy. A hybrid model incorporating Gaussian Process Regression (GPR) in existing physiological models is proposed to guarantee prediction accuracy, physiological insight, and improved interpretability by elucidating the relationship between intestinal components and drug solubility.
The predictive accuracy of the proposed methodologies is tested in different industrial case studies and are further validated for literature benchmarks, paving the way of personalized pharmaceutical industry 5.0 through digital models.
Keywords: Bioreactor modeling, Feed forward neural network, Hybrid semiparametric model, Physics-informed neural network, Fed-batch reactor
Abstract
Bioreactors are fundamental to bioprocess technology, yet the complexity of bioreactor systems continues to challenge effective digitalization and optimization. The intricate, dynamic nature of cell cultivation systems, affected by cell population heterogeneity, genetic instability, and intracellular regulatory mechanisms, poses a significant challenge for modeling and control. Mechanistic models based on first-principles equations mimic the system dynamics through known physical/ biological knowledge. However, they are often constrained by high mathematical complexity, extensive parameterization, identifiability problems, and high computational demand. In response to these limitations, hybrid mechanistic/machine learning approaches are gaining traction in bioprocess applications (Pinto et al., 2022). The hybrid model combines machine learning, e.g., feed forward neural networks (FFNN), with the mechanistic model, alleviating their limitations by incorporating prior knowledge into the model structure, thereby reducing data dependency and enabling more efficient process optimization.
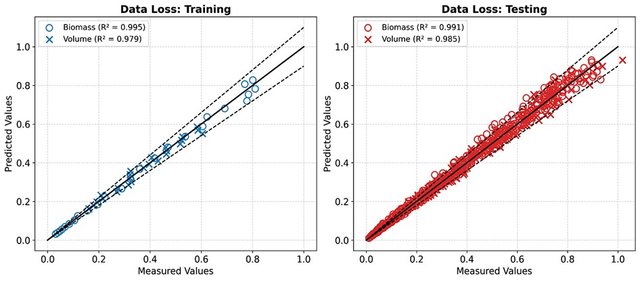
Recently, Physics-Informed Neural Networks (PINN) have emerged as an alternative framework to embed physical laws (expressed through differential equations (ODEs or PDEs)) into neural network models via the training method. This ensures that the neural network models not only fit the available data but also adhere to the underlying governing laws of the system (Raissi et al., 2019). In this study, we propose a tandem FFNN PINN architecture to model a generic bioreactor system. The first network (FFNN-1) parameterizes the bioreactor state variables as a function of time and control inputs, whereas the second network (FFNN-2) parameterizes the reaction kinetics as a function of bioreactor state variables. The training of a PINN involves the simultaneous minimization of data and a physics loss function. The proposed tandem FFNN PINN structure enhances the training convergence and the ability of the model to capture complex nonlinear dynamics typical of bioreactor systems. The proposed PINN framework is benchmarked against a hybrid semiparametric model using several case studies, including a simple microbial logistic growth process and more complex fed-batch bioreactor problems. The models were trained on data from a single batch, and their extrapolation capability was evaluated across 14 testing batches without volume measurement during training, which were governed solely by embedded physical laws. Despite this demanding testing scenario, the PINN model demonstrated high predictive accuracy, achieving a coefficient of determination R2=0.99 for biomass and R2=0.98 for volume (Figure 1). This study shows that PINN are very efficient at capturing bioreactor dynamics with a very low number of data points, demonstrating their ability to generalize to unseen scenarios. There was, however, no definite evidence that PINN overcame hybrid semiparametric modeling for the problems addressed.
REFERENCES
Pinto, J., Mestre, M., Ramos, J., Costa, R. S., Striedner, G., Oliveira, R., 2022. A general deep hybrid model for bioreactor systems: Combining first principles with deep neural networks. Comput. Chem. Eng. 165, 107952.
Raissi, M., Perdikaris, P., Karniadakis, G. E., 2019. Physics-informed neural networks: A deep learning framework for solving forward and inverse problems involving nonlinear partial differential equations. J. Comput. Phys. 378, 686–707.
Keywords: Biopharma 4.0, Deep learning, Physics Informed Neural Networks, Bioreactors, Digital Twin
Abstract
Hybrid modeling combining First-Principles with Machine Learning (ML) is becoming a pivotal methodology for Industry 4.0 enactment. The combination of ML with prior knowledge generally improves the model predictive power and transparency while reducing the amount of data for process development. There is, however, a research gap between emerging machine learning methods and the current practice in bioprocess development [ 1 ].
In this work, we present an automatic control system that makes use of Physics Informed Neural Networks (PINNs) [ 2 ] to act as a digital twin of a natural microbiome in a Sequencing Batch Reactor (SBR) in order to maximize Polyhydroxyalkanoates (PHA) production through the control of 3 different feeds: An Acetate/culture medium solution, an Ammonia/culture medium solution and a culture medium solution (Figure 1).
Firstly, the PINN model was trained with historical data from a Sequencing Batch Reactor (SBR). During this step, deep learning centric approaches, such as the Adaptive Moment Estimation Algorithm (ADAM) were used. After the PINN was trained, a Model Predictive Control (PC) control step was carried out. The PINN model acted as a digital twin of the process to forecast the dynamics of the SBR based on process measurements and 3 decision variables: Feeding rate of acetate, feeding rate of ammonia and feeding rate of medium. Afterwards, based on the forecast, the MPC selects the parameters that maximize the productivity of the next feast and famine cycle (defined as the amount of biomass (VSS) multiplied by the amount of PHA at the end of the feast).
After a total of 14 cycles, corresponding to 3 hydraulic retention times, the PHA has increased from the starting 5 mg/gVSS in 14 g/L of VSS to 226 mg/gVSS in 6.5 g/L of VSS. This was further confirmed via a dedicated accumulation experiment, which resulted in 528 mg/gVSS in 10 g/L of VSS.
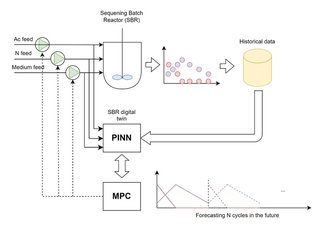
Figure 1. Hybrid control system schematic
References
[ 1 ] Agharafeie, R., Oliveira, R., Ramos, J. R. C., & Mendes, J. M. (2023). Application of hybrid neural models to bioprocesses: A systematic literature review. Authorea Preprints.
[ 2 ] Raissi, M., Perdikaris, P., Karniadakis, G. E., 2019. Physics-informed neural networks: Adeep learning framework for solving forward and inverse problems involving nonlinear partial differential equations. J. Comput. Phys. 378, 686–707. https://doi.org/10.1016/j.jcp.2018.10.045
Keywords: Digital Twin, Hybrid Modeling, Machine Learning, Supercritical Carbon Dioxide Extraction, Process Optimization
Abstract
Supercritical carbon dioxide (ScCO2) extraction is a separation process that presents several advantages over traditional extraction methods of nonpolar solutes, eliminating the need for harmful organic solvents and costly post-processing steps required to remove solvents from extracts. Carbon dioxide is an ideal solvent due to its safety, availability, and cost-effectiveness. Its relatively low critical temperature (304.25 K) allows for the extraction of heat-sensitive substances without degradation (Couto, 2009, Mendes, 2006).
Modeling the ScCO2 extraction process typically involves a combination of intraparticle and macroscopic material balance equations alongside mass transfer laws. A significant challenge in these models lies in defining the relationships between mass transfer coefficients, flow conditions, ScCO2 properties, and the physiochemical characteristics of the target solute. The latter are typically empirical and less reliable, eventually compromising the model's predictive power.
In this study, we developed a hybrid neural network (HNN) model for the ScCO2 extraction of lipids from biomass. The developed HNN combines a feedforward neural network (FFNN) with intraparticle and macroscopic material balance equations, formulated as Partial Differential Equations (PDEs). Particularly, the FFNN is used to model the overall transfer rate of the solute from porous biomass into the bulk as function of biomass microenvironment conditions.
The study used data from ScCO2 extraction experiments (20% for testing and 80% for training the model) to extract lipids from biomass under varying temperatures (313–335 K), pressures (200–500 bar), and ScCO2 flow rates (0.00017–0.0025 kg/sec).
Initially, the extraction column was discretized into multiple levels (3–20), and a mechanistic model was developed using differential equations and empirical mass transfer laws to predict lipids extraction efficiency based on operational parameters. In the next stage, FFNNs were integrated to enhance prediction accuracy. Results showed that increasing the number of discretization elements significantly improves hybrid model predictive accuracy, reducing training and testing errors. The final hybrid model shows high predictive power, eventually supporting a digital twin of the ScCO2 extraction unit. The next step will be to optimize the ScCO2 extraction process.
References
Agharafeie, R., Ramos, J. R. C., Mendes, J. M., & Oliveira, R. M. F. (2023). From Shallow to Deep Bioprocess Hybrid Modeling: Advances and Future Perspectives. Fermentation, 9(10), 1-22. Article 922
Couto, Ricardo M., Joao Fernandes, MDR Gomes Da Silva, and Pedro C. Simoes. "Supercritical fluid extraction of lipids from spent coffee grounds." The Journal of Supercritical Fluids 51, no. 2 (2009): 159-166.
Mendes, R.L., Reis, A.D., Palavra, A.F. (2006). Supercritical CO2 extraction of c-linolenic acid and other lipids from Arthrospira (Spirulina)maxima: comparison with organic solvent extraction, Food Chem. 99, 57–63.
Five samples soils from agricultural farms of Manica province (Mozambique), two of Manica and three of Sussundenga district, were collected by random sampling. The random sampling was done in a zigzag manner in a four years period, from 2021 to 2024 - a total of fifteen samples, five per farm in three campains, were collected [1,2].
Twenty-seven physical-chemical parameters were analysed for all the samples: extractable ions (K, Mg, Ca, Fe, Mg, Zn, Cu and B), exchangeable ions (Na, K, Ca, Mg and Al), cation exchange capacity (CEC), fertility properties of soils (total limestone, active limestone, pHH2O, pHKCl, extractable P, organic carbon, organic matter, N total, N nitrate, conductivity) and texture (sand, clay and slit) [2].
A multivariate analysis design was used to classify the soil samples under research. Cluster analysis (hierarchical and a non-hierarchical K means) was performed in order to evaluate the similitude of the soil samples and the statistically significant variables that contribute to the variability of the model. Also, a two-step cluster and discriminant analysis were performed in order to further explore the results of the initial cluster analysis [3,4].
The non-standardized hierarchical cluster analysis for the five samples at each sampling time resulted in a two-cluster model and for the all the fifteen soil samples along the time a three-cluster model. By a non-hierarchical K means cluster analysis the more stable model, for the five samples at each sampling time, was found with the two clusters model and, for all the fifteen samples along the time, was found for the three-cluster model. The two-step cluster analysis confirmed these previous results. The more and the less important predictor found for each of the five samples for the three sampling times is not the same. For all the fifteen samples the more important predictor found with is the extractable K and the less important predictor is the N nitrate. The discriminant analysis confirms the previous cluster models for the five samples at each sampling time and for all the fifteen samples along the time.
Hybrid modeling has emerged as a cost-effective and time-saving approach for process modeling, significantly advancing model-based process development within the biopharmaceutical industry. By integrating mechanistic and data-driven modeling techniques, hybrid models provide a comprehensive framework that enhances process efficiency and scalability. This combination leverages the strengths of both mechanistic insights and data-centric methods, allowing for more accurate and reliable predictions of process behavior under varying conditions.
A key advantage of hybrid modeling in the pharmaceutical industry is its ability to generate robust models with relatively less data compared to purely data-driven approaches. This is particularly beneficial because data collection in the industry is not only expensive but also time-consuming. Consequently, hybrid models contribute to accelerating the development of new products by minimizing the experimental workload while maintaining model accuracy.
In this work, we explore the application of hybrid models to accelerate the development of critical bioprocesses, particularly focusing on upstream processes for cell and gene therapies, as well as the in vitro transcription (IVT) process used in mRNA production. These bioprocesses often involve complex interactions and dynamic behavior, making them challenging to model using conventional techniques alone.
We present a series of case studies to demonstrate the practical application of hybrid models in optimizing bioprocesses. In the context of cell and gene therapy, hybrid models are used to model cell growth, productivity, and quality attributes, enabling more precise control over critical process parameters. Similarly, in IVT processes, hybrid models facilitate the prediction of yield and quality, streamlining process development and ensuring consistency across production batches.
Our findings underscore the versatility of hybrid modeling in addressing the unique challenges of cell therapy, gene therapy, and mRNA-based processes. By reducing development timelines and improving process performance, hybrid models hold great potential to support the rapid development of advanced biopharmaceutical products.
In the Ceramic district of the cityt of Beira in Mozmabique the drinking water became from treated piped water and from traditional wells without water treatment. Nine sampling sites of water from wells use for human consumation in the Ceramic district near a cemetery were chosen to do an evaluation of the quality of the water of the wells and check the similitudes of the water of different wells. The sampling of water from wells was done by convenience sampling attending to localization of the wells. Nine sampling sites were chosen in order to evaluate the quality of the water and check their similitude in terms of the physical-chemical and microbiological parameters. The nine collected samples were then placed in the fridge [1,2].
The sampled water was then classified attending to the principal physico-chemical and microbiological analytical parameters. The physico-chemical parameters analysed were the pH, conductivity, electrical conductivity (EC), temperature, ammonia, calcium, magnesium, iron, nitrate, nitrite, sulphate, phosphate, chemical oxygen demand (COD), silica, alkalinity, copper, barium, cadmium, cobalt, nickel, zinc, ethilbenzene, styrene, chloromethane, di chloromethane, trichloromethane, chloroethane, biphenyl, tribromomethane, total hydrocarbons C10-C12, bromodichloromethane and dibromochloromethane concentrations. The microbiological parameters analysed are the most probable number (MPN) of fecal and total coliforms.
A classical chemometric cluster analysis was performed in order to evaluate the water samples and the more statistically significant variables that contribute to the variability of the model. A hierarchical and non-hierarchical K means cluster analysis were initially performed in order to evaluate the possible number of cluster and the more statistically significative variables that influence the cluster analysis. Also a two-step cluster analysis was also performed in order to more deeply understand the results of the initial cluster analysis [3,4].
By a non-standardized hierarchical cluster analysis, a two clusters model, a first cluster with two samples and a second cluster with five samples, or a three clusters model, a first cluster and a second cluster with two samples and a third cluster with five samples, seems to be the more adequate cluster models. The same results are found using the method based in the linkage between groups and the Ward’s method as cluster models. A more stable model with two clusters with a great number of statistically significative variables was found with the two clusters model. The two-step cluster analysis allows to confirm that the two-cluster model is the most adequate model. Also, by two-step cluster analysis, and for the two-cluster model, the more important predictor found is the concentration of ammonia and the less important predictor is the pH.
Spectroscopic technologies have seen a substantial rise in applications in recent years, particularly in the monitoring of cell cultures. In addition to this established use, novel applications are emerging, such as the utilization of spectroscopic techniques in microbial fermentations. Alongside these advancements, innovative methods are being developed to create calibration models that effectively correlate spectral measurements with reference concentrations of specific species. Despite these promising developments, the adoption of spectral technologies, especially among smaller companies, remains limited. This is primarily due to challenges associated with data management, as well as the need for specialized knowledge in data analytics and modeling, which can pose a barrier for non-experts.
In this contribution, we present a streamlined approach to calibration model development by employing Bayesian optimization for hyperparameter tuning. This advanced technique enables the automatic selection of data treatment methods, preprocessing techniques, and modeling algorithms, significantly reducing the effort required to develop robust and reliable calibration models. By automating key and complicated aspects of the process, Bayesian optimization empowers users who may lack specialized data analysis skills, thus lowering the entry barrier for adopting spectroscopic methods.
Furthermore, we demonstrate the advantages of implementing this approach on a cloud-based platform, where data is consistently contextualized and made accessible to various stakeholders. Such a platform fosters collaboration among individuals with diverse expertise, promoting more integrated and efficient model development workflows.
Our findings emphasize the potential of Bayesian optimization as a powerful tool for simplifying and democratizing the use of spectroscopic technologies. By enabling more efficient calibration model development and supporting online monitoring, this approach can significantly accelerate the adoption of spectroscopic methods in both research and industrial settings.
In the rapidly evolving field of pharmaceutical bioprocess development, the application of transfer learning techniques presents a transformative opportunity to enhance model performance and streamline process development activities. One of the key challenges in bioprocess development is the high cost and time associated with generating new experimental data. Transfer learning offers a compelling solution by enabling models to leverage existing data from previously established processes. This reuse of historical data is particularly valuable in the pharmaceutical industry, where product development often involves lengthy experimentation and significant resource investment.
The study investigates the efficacy of transfer learning in leveraging historical process data to inform the development of models for new products. We demonstrate that transfer learning significantly improves the predictive accuracy of process models when applied to process data of unseen products. Our results indicate that utilizing knowledge from previously established processes allows models to leverage process information that is common for both the historical and the new product being studied. This leads to a more efficient calibration of models, significantly reducing the number of required experiments while maintaining comparable performance to traditional Design of Experiments (DoE) approaches. This strategy is especially useful when research for the new product is still in an early phase, where data is scarce
This research highlights the potential of transfer learning as a powerful tool in bioprocess development, paving the way for more informed decision-making in the pharmaceutical industry, as well as providing a method to accelerate the development of new products.
The production of solid lipid nanoparticles (SLNs) in the pharmaceutical field presents significant challenges, particularly in terms of optimizing drug loading and colloidal properties, as well as enhancing product quality and manufacturing efficiency. This study aims to address these challenges by investigating the transfer of a process guided by Quality by Design (QbD) principles. Specifically, the transition from hot high-pressure homogenization (hot-HPH) to microfluidic production was explored, since hot-HPH is often associated with high energy and time consumption, limited scalability, and potential batch-to-batch variability, all of which can compromise therapeutic performance. In contrast, switching to microfluidics, driven by QbD, offers improved control, scalability, and efficiency, enhancing both product quality and therapeutic outcomes (1).
QbD provides a structured and systematic approach to understanding and controlling critical material attributes (CMAs) and critical process parameters (CPPs), which are essential to optimizing critical quality attributes (CQAs) in SLNs production. By integrating QbD principles, all relevant parameters are evaluated simultaneously and collectively, enabling a more effective optimization and ensuring a consistent, reproducible product quality.
A key component of this study is the use of failure mode, effects, and criticality analysis (FMECA) as a comprehensive risk assessment tool. FMECA is employed to systematically identify potential failure modes, evaluate their impacts, and prioritize associated risks using the risk priority number (RPN). This analysis focuses on the CPPs that could influence the CQAs, and will be conducted for both microfluidic and hot-HPH processes to allow an in-depth comparison of the transfer process (2).
By integrating QbD principles with FMECA, this study highlights the value of these tools in embedding quality into the manufacturing process and promoting continuous improvement. This integrated approach not only supports robust process optimization but also facilitates a seamless transition between different production technologies, ensuring a high product quality and process efficiency. Ultimately, this approach provides a comprehensive framework for optimizing complex SLNs production processes, making it easier to produce, compare, and transfer between technologies such as HPH and microfluidics.
References
1. Figueiredo J, Mendes M, Pais A, Sousa J, Vitorino C. Microfluidics-on-a-chip for designing celecoxib-based amorphous solid dispersions: when the process shapes the product. Drug Deliv Transl Res. 2025;15(2):732–52.
2. Mendes M, Branco F, Vitorino R, Sousa J, Pais A, Vitorino C. A two-pronged approach against glioblastoma: drug repurposing and nanoformulation design for in situ-controlled release. Drug Deliv Transl Res. 2023;13(12):3169–91.
End-to-End (E2E) models and Digital Twins in the pharmaceutical industry enhance efficiency, improve decision-making, allow for real-time monitoring, optimization, predictive analytics, and ultimately strengthen quality control and reduce costs. A key component of E2E models is the use of Monte Carlo simulations to capture uncertainties and variability within complex processes.
Typically, E2E models rely on linear regression models due to their simplicity, ease of implementation, and high computational speed. This makes them particularly suitable for Monte Carlo simulations, which require numerous iterations to achieve statistically meaningful results. However, linear regression models may be too simplistic for accurately representing certain dynamic aspects of pharmaceutical processes. In such cases, more complex models — such as those based on Ordinary Differential Equations (ODEs) and Partial Differential Equations (PDEs) — are necessary to provide a more precise and mechanistic understanding of process dynamics.
Despite their advantages, ODE- and PDE-based models present significant computational challenges. Unlike linear regression models, solving differential equations is computationally intensive, leading to long processing times. When incorporated into Monte Carlo simulations, this results in extremely prolonged computation durations, particularly as the number of required simulations increases. Additionally, these computational demands make real-time applications nearly impossible or require compromises in prediction accuracy.
To address these limitations while maintaining the benefits of ODE and PDE solvers in Monte Carlo simulations, we have developed surrogate models leveraging Physics-Informed AI. These surrogate models significantly accelerate simulations while preserving the accuracy of mechanistic models. By integrating physics-based constraints into AI-driven approximations, we enable more efficient uncertainty quantification and process optimization without the computational burden of traditional solvers.
Our approach enhances the applicability of E2E models and Digital Twins in pharmaceutical process modeling, paving the way for faster, more accurate decision-making and real-time process optimization. This advancement has the potential to revolutionize drug development and manufacturing by making sophisticated modeling techniques more accessible and practical for industrial applications.
Despite advancements in Systems Biology, developing purely ODE-based mechanistic models remains challenging due to incomplete knowledge of parameters or computational inefficiencies. In such cases, hybrid and data-driven approaches provide viable alternatives. To facilitate seamless simulation and analysis alongside classical ODE-based models, it is advantageous to encode data-driven models in the Systems Biology Markup Language (SBML).
This work presents a framework for encoding neural networks of various architectures in SBML, enabling their integration into existing computational biology tools. We implemented feed-forward and Elman recurrent neural networks (RNNs) with Tanh and ReLU activation functions. Our approach is demonstrated using two well-known case studies: (1) the Escherichia coli threonine synthesis model and (2) protein synthesis in a fed-batch bioreactor.
To validate our framework, we simulated the models in COPASI, a widely used tool for SBML-based simulations. While the Tanh function is natively supported in SBML, it often results in slow convergence during training. We explored the introduction of alternative activation functions, such as ReLU, via user-defined functions. Additionally, we examined the advantages of Elman RNNs over feed-forward networks, particularly for time-series modeling. Our results indicate that Elman RNNs outperform feed-forward networks in cases where data is limited and the system evolves rapidly, as observed in the bioreactor case study. Recurrence in RNNs was implemented using SBML events.
Further analysis revealed that decomposing mathematical expressions—by defining each neural network node as an SBML species or parameter—significantly reduces simulation time in COPASI compared to models where assignment rules are applied only to outputs. This effect becomes particularly pronounced in larger models.
Overall, our findings highlight the feasibility of encoding neural networks in SBML, which can be useful for further development of hybrid models.
REFERENCES
Pinto, J., Mestre, M., Ramos, J., Costa, R. S., Striedner, G., Oliveira, R., 2022. A general deep hybrid model for bioreactor systems: Combining first principles with deep neural networks. Comput. Chem. Eng. 165, 107952. https://doi.org/10.1016/j.compchemeng.2022.107952
Introduction: The blood-brain barrier (BBB) severely restricts the passage of drugs into the brain, posing a significant challenge in treating central nervous system disorders such as glioblastoma (GBM). Therefore, there is an urgent need for advanced in vitro models that accurately characterize both BBB permeability and GBM behavior [1]. The evolution from two-dimensional (2D) to three-dimensional (3D) models, especially those incorporating dynamic conditions, is critical to enhancing our understanding and predictive capabilities regarding BBB permeability and GBM treatment [2,3]. In this study, we used 3D printing technology to develop a 3D model of the BBB and GBM microenvironment.
Materials and methods: The model is designed to replicate the complex architecture and dynamic conditions of the BBB and tumor tissue. It features a cylindrical structure with a core composed of a gelatin methacrylate (GelMA)-alginate bioink containing U87 glioblastoma (GBM) cells, microglia (HCM3), and astrocytes. Surrounding this core, a GelMA-fibrinogen bioink incorporating human brain microvascular endothelial cells (HBMECs) forms the perimeter.
Results: A risk-based analysis, using an Ishikawa diagram and a risk estimation matrix, identified potential sources of variability in the model. Principal component analysis assessed the impact of process variables, including nozzle type, pressure, temperature, and crosslinking, on critical quality attributes (CQAs). These CQAs encompassed printability, structural integrity, stiffness, porosity, and cell viability, with cell viability being the primary focus.
Conclusion: 3D bioprinting holds great potential for creating physiologically relevant and reproducible models that enhance our understanding of GBM biology. It may also act as a dependable platform for evaluating the effectiveness of drugs and nanoparticles, thus promoting personalized therapies.
References:
[1] J.P. Straehla, C. Hajal, H.C. Safford, G.S. Offeddu, N. Boehnke, T.G. Dacoba, J. Wyckoff, R.D. Kamm, P.T. Hammond, A predictive microfluidic model of human glioblastoma to assess trafficking of blood–brain barrier-penetrant nanoparticles, Proc Natl Acad Sci U S A 119 (2022). https://doi.org/10.1073/PNAS.2118697119/SUPPL_FILE/PNAS.2118697119.SD01.XLSX.
[2] M. Tang, J.N.Rich, S. Chen, M. Tang, S. Chen, J.N. Rich, Biomaterials and 3D Bioprinting Strategies to Model Glioblastoma and the Blood–Brain Barrier, Advanced Materials 33 (2021) 2004776. https://doi.org/10.1002/ADMA.202004776.
[3 ]F. Branco, J. Cunha, M. Mendes, J. J. Sousa, C. Vitorino, 3D Bioprinting Models for Glioblastoma: From Scaffold Design to Therapeutic Application. Adv. Mater. 2025, 2501994. https://doi.org/10.1002/adma.202501994
Quality by Design (QbD) is a systematic approach for building quality into a product. The Design Space Profiler in JMP helps solve the fundamental QbD problem of determining an optimal operating region that assures quality as defined by specifications associated with Critical Quality Attributes (CQAs) while still maintaining flexibility in production.
We will discuss a simulation approach to solving this difficult problem and demonstrate this approach using JMP’s Design Space Profiler. The Design Space Profiler is part of the Prediction Profiler in JMP and provides an interactive way to find and explore the Design Space.
All demo content will be shared with the participants, while the academic license 'JMP Student Edition' is available free of charge.
Establishing multivariate specification regions for selecting raw material lots entering a customer’s plant is crucial for ensuring smooth operations and consistently achieving final product quality targets. Moreover, these regions guide the selection of suppliers. By meeting these specifications, suppliers contribute to customer satisfaction, which can, in turn, enhance market share. Latent Variable Methods, such as Partial Least Squares regression (PLS) and the more recent Sequential Multi-block PLS (SMB-PLS), have proven to be effective data-driven approaches for defining multivariate specification regions. These methods model the relationships between raw material properties (Critical Material Attributes – CMA), process variables (Critical Process Parameters – CPP), and final product quality (Critical Quality Attributes – CQA), enabling the identification of a lower-dimensional latent variable subspace that captures quality-relevant variations introduced by raw materials and process conditions. This subspace is central to the methodology.
Within this latent variable space, several statistical limits are defined to ensure final product quality and guarantee data compliance with the latent variable model, collectively forming the multivariate specification region. After providing historical context on the early development of these methods, this presentation will cover several key topics. These include the data requirements for constructing latent variable models and how to organize them into distinct blocks. The techniques used to define the limits (in terms of shape and size) within the latent variable subspace—via direct mapping and latent variable model inversion—will be discussed, along with methods for addressing uncertainties. Particular attention will be given to how process variations generate different scenarios and approaches for establishing meaningful specification regions. Case studies using both simulated and industrial data will illustrate these methods. It will be demonstrated that the proposed framework aligns with the principles of Quality by Design (QbD), with notable similarities to the Design Space (DS) concept. The presentation will conclude by exploring potential future developments, including the establishment of multivariate process capability indices based on specification regions.
Inception for Petroleum Analysis (IPA) [1] is a deep convolutional network inspired from state-of-the-art computer vision architectures. IPA showed improved performance, compared to PLS, without depending on complex pre-processing operations thanks to its several computational blocks. The network begins with three stacked convolutions, followed by a multi-branch module consisting of four different paths that are concatenated. The model learns complementary but complex, internal representations. eXplainable Artificial Intelligence (XAI), a hot topic in DL can bring understanding on such complex models and help to envisage deep networks as robust and performant alternatives to traditional PLS. Two studies using gradient-weighted class activation mapping (Grad-CAM) [2,3], already have been applied to shallow CNNs, extending these methods seems fundamental for DL to earn the confidence of the chemometrics community. The application of XAI [4,5] for NIR analysis can be greatly improved and is crucial for many practical applications such as agriculture, environmental monitoring and oil characterization.
The new framework we proposed is built on a two-step analysis. Explainability has been positioned at several points within the architecture to demonstrate the complementarity of each computational blocks. First, it was crucial to understand the features globally influential for the model, considering the chemical aspect necessary for the property of interest to impartially judge the quality of the deep CNN. Finally, layer-wise explanations was carried out to determine the contribution of each layer to the overall model performance.
Feature importance showed that both models put importance on the same spectral regions, while the PLS’s interest is restricted and diffuse, IPA’s interest is smoother and more moderate. The layer-wise explanations demonstrated that the performance of IPA comes from its complementary multi-branch layers inspired by the Inception model. It has been demonstrated, without any prior knowledge or pre-processing, that the network focus on relevant spectral region in terms of physico-chemical in relation to the full range of cetane number. In addition, this XAI methodology illustrated that these types of models are not only performant but also more robust than shallower CNNs and PLS. Specifically, IPA better identifies the important spectral regions in relation to the molecular composition of the product that influence the cetane number, such as aromatic content as well as the length of the hydrocarbons molecules.
References:
[1] Haffner F. et al.; IPA: A deep CNN based on Inception for Petroleum Analysis. Fuel 2025, 379, 133016.
[2] Yang J. et al.; An interpretable deep learning approach for calibration transfer among multiple near-infrared instruments. Computers and Electronics in Agriculture 2022, 192, 106584.
[3] Pasos D.; Mishra P.; An automated deep learning pipeline based on advanced optimisations for leveraging spectral classification modelling. Chemometrics and Intelligent Laboratory Systems 2021, 215, 104354.
[4] Lundberg S. et al.; Explainable machine-learning predictions for the prevention of hypoxaemia during surgery. Nature biomedical engineering 2018, 10, 749-760.
[5] Selvaraju, R. et al.; Grad-CAM: Visual Explanations from Deep Networks via Gradient-based Localization. International Journal of Computer Vision 2020, 128, 336-359.
In artificial intelligence (AI), the complexity of many models and processes often surpasses human interpretability, making it challenging to understand why a specific prediction is made. This lack of transparency is particularly problematic in critical fields like healthcare, where trust in a model's predictions is paramount. As a result, the explainability of machine learning (ML) and other complex models has become a key area of focus.
Efforts to improve model interpretability often involve experimenting with AI systems and approximating their behavior through simpler mechanisms. However, these procedures can be resource-intensive. Optimal design of experiments, which seeks to maximize the information obtained from a limited number of observations, offers promising methods for improving the efficiency of these interpretability techniques.
To demonstrate this potential, we explore Local Interpretable Model-agnostic Explanations (LIME), a widely used method introduced by Ribeiro et al. 2016. LIME provides explanations by generating new data points near the instance of interest and passing them through the model. While effective, this process can be computationally expensive, especially when predictions are costly or require many samples.
LIME is highly versatile and can be applied to a wide range of models and datasets. In this work, we focus on models involving tabular data, regression tasks, and linear models as interpretable local approximations.
By utilizing optimal design of experiments' techniques, we reduce the number of function evaluations of the complex model, thereby reducing the computational effort of LIME by a significant amount. We consider this modified version of LIME to be energy-efficient or ``green''.
Convolutional Neural Networks (CNNs) have been increasingly used to build NIR based chemometric models with applications ranging from chemical sample analysis to food quality control. In the latter, NIR spectroscopy combined with CNNs enable rapid, non-destructive SOTA predictions of important quality parameters such as dry matter content in fruit [1].
The lack of a standard CNN architecture optimized for spectral data, makes the use of these algorithms a non-trivial task. In this work we approach this problem by performing a joint neural architecture search (NAS) and hyperparameter optimization (HPO) to find an appropriate architecture for the task of dry matter (DM) prediction in fruit. DM is a critical quality indicator, influencing nutritional value, shelf life, and overall consumer acceptance. To improve model robustness, we used a dataset that includes four fruit types (apples, kiwis, mangoes, and pears). Variations in fruit morphology, cultivar, and origin introduces substantial spectral variability, posing challenges for traditional chemometric methods. The multi-fruit spectra were acquired with the same spectrometer model (but different devices) and pre-processed using the second derivative to enhance feature extraction. By training a deep learning model on a multi-fruit dataset we aimed at improving its generalization capabilities.
Our joint NAS and HPO process uses Bayesian optimization via the Tree-structured Parzen Estimator (TPE) [2] where different CNN configurations are evaluated for DM prediction. Models were trained under different data distribution assumptions, initialization strategies and from shallow networks, with a single convolutional layer and one dense layer, to more complex configurations featuring multiple convolutional and dense layers. The HPO included the number and size of convolutional filters, dropout rates, L2 regularization strengths and number of units in dense layers using a cross-validation strategy and aiming at minimizing RMSE. Furthermore, the integration of dual-task models that performed both regression (for DM prediction) and classification (for fruit type identification) allowed the CNNs to leverage shared information between tasks. This allowed to train CNN global models (trained on all fruit types) that performed better than individual PLS models (single fruit models) and global and Locally Weighted PLS models [3].
A second study [4] addressed one of the main criticisms deep learning models face in chemometrics, i.e. their “black box” nature. We show that the use of model-agnostic explainability techniques such as Regression Coefficients [5], LIME [6], and SHAP [7] can be used to reliably identify the spectral bands that most strongly influenced the CNNs’ predictions. Our analysis demonstrated that the key wavelengths identified by the CNNs coincided with those recognized by classical methods like PLS VIP scores and theoretical expectations. Moreover, we show that the CNN leans towards using the domain invariant features across fruit samples (akin di-CovSel [8]). An in-depth analysis of the learned convolution kernels also helps us understand that CNNs perform a type of data driven preprocessing for different samples in the dataset.
We are currently extending the lessons learned here to other datasets to further validate out methodology.
Design of experiments is one of the main Quality by Design (QbD) tools within the process industry.
However, "classic" DoE is increasingly challenged by modern techniques such as Bayesian Optimization and Active Learning.
These innovative methods are promoted as faster and more intuitive, offering greater flexibility in experimentation.
In this talk, I will provide a direct comparison of classic DoE and Bayesian Optimization performance in typical R&D and process optimization settings. I'll consider experimental effort (the number of experiments required), practical aspects, and the quality of information generated. This comparison will help determine when each approach is most appropriate.
Spoiler: My impression is that the perception of classic DoE as requiring extensive experimental effort is partly influenced by the philosophy of its practitioners. Therefore, I advocate for a "back-to-the-roots" approach: a strongly risk-based deployment of sequential DoE. I look forward to discussing these points with you.
Bayesian Optimization (BO) has been recently shown as an efficient method for data-driven optimization of expensive and unknown functions. BO relies on a probabilistic surrogate model, commonly a Gaussian Process (GP), and an auxiliary acquisition function that balances exploration and exploitation for a goal-oriented experimental design, with the aim of finding the global optimum under a reduced experimental budget.
Before BO gained popularity, traditional statistical Design of Experiments (DOE) methods were commonly used to tackle such problems, such as, but not limited to, classic full/fractional factorial, response surface designs (central composite or Box-Behnken), and optimal designs. There are also other more recent developed methods such as Definitive Screening Designs or Orthogonal Minimally Aliased Response Surface (OMARS) designs. In contrast to BO, these designs are generally focused on the estimation of a quadratic response surface model and are mainly static in nature, although several rounds of experiments are typically recommended in practice where previous rounds’ results are incorporated before the next round of experiments is determined. Nonetheless, there is a wide body of work, both theoretical and application driven, with success stories in different fields.
In this work, we consider a real-world chemical synthesis problem, where the goal is maximizing the reaction yield by manipulating a set of continuous and categorical variables, with a total of 22 variables, after One-Hot-Encoding (OHE). This complex case study provides an appropriate scenario for comparing traditional and adaptive experimental design approaches for optimization.
For the surrogate model in BO, we evaluate both using the OHE approach and the use of chemical descriptors as informative features of the categorical variables. Due to a high-dimensional design space, we also consider the use of Sparse Axis-Aligned Subspace Bayesian Optimization (SAASBO), a fully Bayesian approach which places strong sparsity priors on the GP model to avoid overfitting and relies on Markov Chain Monte Carlo for inference.
We then compare the BO approaches with a sequential statistical DOE approach, where an initial design is performed for variable screening, followed by a second and more efficient design on the variables deemed most important . The advantages and disadvantages of the different methods are highlighted in terms of implementation difficulty, rate of convergence to the optimum as well as model fit quality.
Carefully designing experiments is crucial for gaining a deeper understanding of process behaviour. Design of Experiments (DOE) is a well-established active learning methodology with an extensive track record of solid contributions to research and industry in various areas, including screening, modelling, optimisation, specification matching, and robust design. Based on a reduced set of assumptions regarding process behaviour, DOE proposes an experimentation plan for estimating a parametric model, upon which follow-up experiments may be suggested in a sequential and iterative manner until the intended goal is achieved.
More recently, other methodologies have emerged and are being increasingly tested and explored, primarily focusing on optimization for a specific goal. Among these, Bayesian Optimisation (BO) stands out, gaining recognition and interest for its simplicity and effectiveness in certain test scenarios. Unlike DOE, where modelling and optimisation are treated sequentially as distinct tasks, BO aims to enhance sampling efficiency for optimisation, relaxing the need for an explicit modelling process[1]. Linear least squares models are central to DOE, which creates challenges and awkwardness to avoid design matrix singularities. These problems largely disappear in BO with mixture and constrained effects because the model is based on spatial correlations rather than design matrices. The question that practitioners face now is which methodology to adopt when the goal is optimisation. Despite the numerous papers published, the vast majority address small toy examples. Although these examples are interesting and useful for elucidating the properties of the methodologies, they hardly provide a suitable basis for inference in real systems. In summary, there is currently a scarcity of comprehensive studies and practical case studies that compare these two approaches and assess their relative efficiency in realistic and challenging scenarios.
To promote discussion and gain further insights into their relative merits, we have adopted a realistic simulated scenario as a test bed to compare the two approaches for active learning (DOE vs BO). The system regards the synthesis of lipid nanoparticles (LNPs)[2], which is currently an important process for the development of modern drug delivery systems. Both approaches were applied and comparably tested for their efficiency in achieving optimal values for the examined responses (“potency” and average “size” of LNPs).
Waste lubricant oil (WLO) is a hazardous residue that requires proper management. Among the options available, regeneration is the preferred approach to promote a sustainable circular economy. However, WLO regeneration is only viable if the WLO does not coagulate during processing as it can cause operational problems and possibly lead to a premature shutdown of the process for cleaning and maintenance. To mitigate this risk, a laboratory analysis using an alkaline treatment is currently used to assess the coagulation potential of the WLO before it enters the regeneration process. Nevertheless, this laboratory test is time-consuming, carries several safety risks, and its result is subjective, depending on visual interpretation by the analyst.
In this work, a rapid and robust approach to predicting the coagulation potential of WLOs using multiblock machine learning with near-infrared (NIR) and mid-infrared (MIR) spectroscopy data is proposed. Classification models were trained using interval partial least squares (iPLS) (Nørgaard et al., 2000), PLS for discriminant analysis (Barker & Rayens, 2003), and convolutional neural networks (CNN) (Yang et al., 2019). As the performance of the multiblock PLS models depends on the specific pre-processing and scaling of each spectral block, an exhaustive search over 1755 combinations of pre-processing, scaling, and modelling methods was performed automatically using the AutoML framework called SS-DAC (Rato & Reis, 2019). For reference, the single-block models, for both PLS and CNN methodologies, were also included in the study.
The single-block models indicated clear performance differences, with the MIR block exhibiting superior predictive performance than the NIR block. The best MIR single-block model using PLS achieved a classification accuracy of 0.90, while the MIR single-block CNN model attained an accuracy of 0.94. In contrast, the NIR single-block models showed significantly lower predictive performances, with the best NIR single-block model reaching a classification accuracy of only 0.47. Nevertheless, the integration of both spectral blocks led to an overall improvement in the multiblock PLS models, which attained an accuracy of 0.94. This enhancement is primarily attributed to an increase in the correct classification of WLOs that do not coagulate, which are the most critical ones for the process operation. In contrast, the multiblock CNN model had a decrease in accuracy to 0.87 compared to its single-block counterpart, suggesting that the inclusion of non-informative data negatively impacted its performance. Overall, these findings demonstrate that the combined use of NIR and MIR spectra can improve the capabilities of prediction models compared to their individual use in single-block models.
Keywords: Waste lubricating oil; Multiblock modeling; Classification; Partial Least Squares; Deep Learning
Real-time monitoring of chemical processes is key for optimizing yield, preventing out of specification product, and improving overall process efficiency. Process analytical technology (PAT) tools, such as near-infrared and infrared (IR) spectroscopies, provide a real-time window into chemical processes, enabling non-destructive monitoring of analyte concentrations, and reducing dependance on labor-intensive off-line analytical methods (e.g., gas chromatography). Nevertheless, there are cases where the application of spectroscopy alone can be challenging, e.g., when analyte concentrations reach the method limit of quantification (LOQ). Moreover, PAT alone cannot predict future time steps of the process.
The present work showcases the application of in-line monitoring coupled with a hybrid modeling approach for chemical reaction. An IR probe is submerged in solution within the reactor and a Partial Least Squares (PLS) model developed to predict in real time the concentration of the limiting reagent directly from IR spectra. The PAT is then combined with the process mechanistic knowledge in a Kalman filter, which has previously proven to reduce predictions uncertainty [1]. Here, a second-order reaction kinetic model with two reagents forming one insoluble product was assumed. No additional calibration effort is necessary in this case because the reaction kinetic model is continuously reparametrized during the reaction, based on the PAT predictions. The predictions of these two models were combined with the application of the Kalman filter to optimize estimates.
The application was successfully implemented at the laboratory scale where the hybrid model was able to capture the physics of the reaction, providing end-point determination with higher degrees of accuracy. This strategy not only reduced the PAT LOQ and improved production scheduling but also reduced turnaround time and analytical costs. Additionally, it eliminated the need for manual sampling, mitigating the risks of operator exposure to the material.
References
[1] D. Krämer and R. King, “spectroscopy and a sigma-point Kalman filter,” J. Process Control, 2017.
Monitoring formulation quality during Continuous Direct Compression (CDC) and therefore remaining within product specifications is complex and cannot easily be inferred from process measurements. Process Analytical Technology (PAT) sensors allow in-line process monitoring and control of Critical Quality Attributes (CQAs), reducing the time and effort required for both sampling and off-line analysis.
This work delves into optimising PAT for CDC, focusing on integrating Near-Infrared (NIR) spectroscopy with the Feed Frame Simulator (FFSIM). The project aims to enhance the accuracy and efficiency of the sensors and models, ensuring consistent product quality even for low-dose formulations.
Key components include optimising the acquisition parameters of the SentroPAT FO NIR spectrometer, such as the integration time and number of averages, as well as utilising the FFSIM for dynamic data collection and model development. This involves Principal Component Analysis (PCA) and Partial Least Squares (PLS) regression. By standardising data analysis methodologies and developing an integrated Real Time Release Testing (RTRT) strategy, the project seeks to address current challenges in PAT applications.
Future developments will explore the simultaneous use of multiple PAT instruments, including Raman spectroscopy and Light-Induced Fluorescence (LIF) spectroscopy, to further enhance process control. The ultimate goal is to create a robust, transferable PAT strategy for the CDC line, facilitating standardised practices across different sites and processes. This will provide insights for the development of new continuous manufacturing control strategies for low dose formulations.
Online analysis has been widely developed to monitor the chemistry or the physics on batch or continuous processes. One of the major issues concerns the sampling part to integrate the analytical solution into the process. Optical spectroscopy is one of the most used technologies as it can be implemented directly inline and does not necessary required a sampling loop to adapt the process to the analyzer. The main advantage is the possibility to use probes or flow cells that can be immerged directly into the process. A wide range of probes exists to be able to fit with the process conditions like the temperature, pressure, or rotation speed. The possibility of using optical fibers allows to deport the analyzer from the processes to protect the sensitive part of it.
In the case of reactive extrusion processes, online analysis is not commonly used as the constraints have some difference from the classical batch or continuous processes. Harsh conditions like high temperature and pressure, melt product with high viscosity make sampling part challenging. Optical spectroscopy can monitor most of the reaction done inside an extruder and the objective here is to present how optical spectroscopy probes can be implemented directly inline. Reactive extrusion like grafting onto a polymer, depolymerization or even homogeneous mixing of polymers are presented to demonstrate its interest.
Online analysis into extruder can be used to do optimization of the process conditions to find the best quality product. An approach using near infrared spectroscopy, chemometrics tools like the PCA (Principal Component Analysis) and a design of experiments based on Bayesian optimization is also presented here. This approach allows to put in place self-optimization of the processes.
In the Chemical Manufacturing Industry, a diverse array of sensor technologies and data collection methods provide valuable insights into monitoring physical and chemical phenomena, equipment status, process conditions, raw material attributes, product quality, emissions, and logistics. Despite the extensive use of sensors, critical process information such as leaks, corrosion, and insulation degradation often goes undetected, often hidden by control actions.
To address this gap, alternative information sources are essential for gaining insights into the state and health of industrial processes. One promising alternative is industrial text data found in reports, alarm tags, quality notes, and process memorandums. Although numerous text processing methods exist, their application in the Chemical Manufacturing Industry remains underexplored. This talk cover the efficacy of text processing techniques for information retrieval from industrial text data.
Industrial text data presents unique challenges for Natural Language Processing (NLP), including specific vocabulary, incomplete information, sparse domain coverage, and complex underlying phenomena. To overcome these challenges, fine-tuning NLP and Machine Learning (ML) models for specific production contexts is a potential solution. This approach requires a high-quality dataset representative of the targeted manufacturing process but poses limitations in generalizing to other processes and sites.
Generative AI (GenAI) models offer a promising solution for industrial text analysis and targeted information extraction. By serving as foundational models, GenAI can support the analysis of industrial text data. This talk will present the journey of industrial text analysis, covering from most basic concepts such as word counting to advanced ones like GenAI applications, highlighting the potential of the industrial text data analysis and including at least one case study.
Background
Monoclonal antibodies (mAbs) are highly specific proteins used in personalized therapeutics, with applications ranging from cancer treatment to autoimmune disease management. In this study, we focus on the production of 86 monoclonal antibody (mAb) molecules, each potentially having unique production characteristics.
Due to the confidential nature of proprietary data in the biopharma industry, obtaining experimental data for all 86 mAb molecules is challenging. Many companies are unwilling to share such data, and this lack of transparency limits the ability to develop generalized models for mAb production processes. Therefore, alternative approaches that can simulate production processes and predict mAb yield are essential to advance our understanding and optimize production methods.
Aims and Objectives
This study aims to develop methods that can enhance the transfer of knowledge across various process development campaigns in the biopharma industry. By leveraging available data and creating synthetic datasets where necessary, we aim to fill gaps in knowledge regarding monoclonal antibody production and improve predictive capabilities. Specifically, the study aims to:
1. Develop dynamic simulation models for mAb production.
2. Use synthetic data generation to compensate for the lack of experimental data.
3. Create a hybrid modeling approach that combines empirical data with knowledge-driven structures to simulate the production processes more accurately.
Methods
The primary tool used for developing the simulation model is Python, with the scipy.integrate module for solving ordinary differential equations (ODEs). The model focuses on defining kinetics constants and rates that govern the production of mAbs and use of aminoacids within the system. The approach involves solving ODEs that describe the behavior of the system over time.
Furthermore, a simple use of Logistic Regression from the sklearn module is employed to assign low titer and high titer to the different mAbs based on factors such as the frequency of amino acids in their sequences and their overall production characteristics.
Results
The results of this study include the development of Python code capable of reading commercial mAb sequences and simulating the production process. The code also accounts for high titer and low titer mAbs, providing insights into how variations in the amino acid sequence can impact production outcomes.
Furthermore, synthetic datasets were generated to visualize the time-course behavior of species within the system, allowing for the observation of changes in mAb concentration and the related biochemical processes over time. These visualizations provide insights into the dynamics of mAb production and the factors that influence titers in different scenarios.
Future Work
The next step involves using the synthetic data generated in this study to train the hybrid model. This will help to improve the model's predictive accuracy, enabling it to handle real-world, incomplete data scenarios typical in biopharma production. By doing so, we aim to develop a more robust and versatile tool for optimizing monoclonal antibody production processes across various biopharmaceutical platforms.
Through this approach, we hope to contribute to the ongoing efforts in the biopharma industry to enhance the efficiency of mAb production while reducing costs and time to market.